โรคนี้ได้รับแรงบันดาลใจจากความจริงที่ว่าถ้ามันไม่ได้ระบุทารกแรกเกิดในเวลามันเต็มไปด้วยการละเมิดการพัฒนาจิตใจและร่างกายของเขา. Mirsees จะบอกผู้อ่านเกี่ยวกับอาการสาเหตุของการเกิดขึ้นของพยาธิวิทยาและวิธีการวินิจฉัยและการรักษาที่มีอยู่.
phenylketonuria คืออะไร?

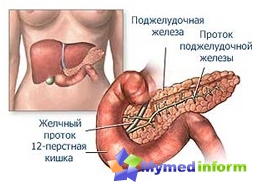
ด้วยโรคที่กำหนดความไม่สามารถของร่างกายได้รับการติดตามถึงวิธีที่ซื่อสัตย์ในการแยกกรดอะมิโนของ Phenylalanine ดังนั้นเนื้อเยื่อจะถูกสะสมโดยสารที่วางยาพิษสมองและระบบประสาท. เด็ก ๆ ดังกล่าวอยู่ในอนาคตที่กำลังพัฒนาจำนวนน้อยในคำอื่น ๆ - ปัญญาอ่อน, งี่เง่าดังนั้นโรคที่ได้รับและคำจำกัดความอื่น ๆ - oligophrenia phenylpyrovinograde. วิธีการวินิจฉัยที่ทันสมัยช่วยให้สามารถระบุโรคตั้งแต่อายุยังน้อยในเด็กดังนั้นเมื่อทำการวัดบางอย่างคุณสามารถเติบโตและให้ความรู้แก่สุขภาพ. บันทึกสมองทารกแรกเกิดจัดการการสังเกตอาหารพิเศษซึ่งจะกล่าวถึงในภายหลัง. เป็นเวลานานโรคที่ศึกษาดร. ตกจากนอร์เวย์ซึ่งอธิบายในปี 1934. สำหรับความพยายามและการมีส่วนร่วมในการแพทย์ของแพทย์นี้โรคนี้ได้รับชื่อที่สอง - โรค fethulling. ในวัยห้าสิบของศตวรรษที่ผ่านมาแพทย์จากอังกฤษทำงานในโรงพยาบาลเด็กพบวิธีการรักษา. และในช่วงปี 1958 ถึงปี 1961 เทคนิค Gastri ได้รับการแนะนำอย่างแข็งขันช่วยให้ทราบถึงเนื้อหาที่เพิ่มขึ้นของ Phenylalanine ในเลือด.
นักวิทยาศาสตร์พบว่าความทุกข์ทรมานจาก FCU แสดงกิจกรรมที่ไม่ถูกต้องหนึ่งในเอนไซม์ตับที่รับผิดชอบการเปลี่ยนแปลงของฟีนิลลานินินไปยังกรดอื่น - ไทโรซีน. จำนวนคนที่เกิดมาพร้อมกับพยาธิสภาพนี้ในประเทศต่าง ๆ ของโลกที่แตกต่างกันอย่างมีนัยสำคัญ. สถิติแสดงให้เห็นว่าในรัสเซียเด็กหนึ่งคนจาก 10,000 คนปรากฏขึ้นกับโรคนี้. ในสหราชอาณาจักรเกิดขึ้นกับ FCU สองเท่า แต่ในแอฟริกาแทบจะไม่มีการเจ็บป่วย. ในเด็กผู้หญิงปัญหาทางพันธุกรรมดังกล่าวจะถูกตรวจพบบ่อยกว่าเด็กผู้ชาย. หากพ่อแม่ทั้งสองมียีนที่ชำรุดเด็กสามารถเกิดได้. ยีนที่ถูกเปลี่ยนพบใน 2% ของคนในขณะที่พวกเขามีสุขภาพดี. แต่ถ้ามีชายคนหนึ่งและผู้หญิงที่มียีนที่มีการกลายพันธุ์และจากนั้นสร้างครอบครัวแล้วความเสี่ยงของการเกิดพวกเขามีเด็กผู้ป่วย 25%. มรดกประเภทนี้เรียกว่า autosomal-recessive.
สาเหตุของการเกิดขึ้น
นักวิทยาศาสตร์จัดสรรสาเหตุต่อไปนี้ที่เพิ่มความเสี่ยงของพยาธิวิทยาที่เด็ก:
- การกลายพันธุ์ของยีนตั้งอยู่บนโครโมโซมที่ 12. สิ่งนี้นำไปสู่การขาดเอนไซม์ Phenylalanine-4-Hydroxylase โดยไม่มีการเปลี่ยนแปลงปกติของ Phenylalanine ในไทโรซีนเป็นไปไม่ได้สิ่งนี้นำไปสู่การสะสมของอนุพันธ์ Phenylalanine อื่น ๆ ที่ส่งผลกระทบต่อเซลล์สมองประสาทและเซลล์.
- การแต่งงานใกล้เคียง.
ในฐานะที่เป็น Phenylketonuria ปรากฏตัวเอง?

ปัจจุบันในศูนย์พันธุศาสตร์คุณสามารถตรวจสอบพิเศษที่จะแสดงว่าผู้ปกครองมีการละเมิดในยีนในกรณีที่มีความสงสัย.
ในรัสเซียในปี 1993 คำสั่งของกระทรวงสาธารณสุขได้ลงนามในปี 1993 ตามที่เด็กทุกคนในโรงพยาบาลคลอดอยู่ภายใต้การคัดกรองทารกแรกเกิดทำให้ฉันสามารถเปิดเผยฟีโนเก็ตก่อนและโรคทางพันธุกรรมอื่น ๆ. สำหรับสิ่งนี้จากส้นเท้าของทารกแรกเกิดสามชั่วโมงหลังจากขั้นตอนการให้อาหารใช้เลือด. หากเด็กอยู่ด้านล่างนี้จะทำในวันที่สี่หลังคลอด แต่ในภายหลังภายหลัง - ในวันที่เจ็ด. จากนั้นรูปแบบการทดสอบที่มีเลือดบาดแผลถูกส่งไปยังห้องปฏิบัติการซึ่งการวิเคราะห์ที่จำเป็นในระหว่างวัน. ผลลัพธ์ได้รับการแก้ไขในการ์ดเด็กวัยหัดเดินที่มีตราประทับพิเศษ. และหากมีความสงสัยของโรคทางพันธุกรรมนี้เด็กและผู้ปกครองให้การอ้างอิงถึงการตรวจสอบเพิ่มเติมในศูนย์การแพทย์และทางพันธุกรรม.
การวิจัยเพิ่มเติมดำเนินการ:
- ในเซรั่มและคราบแห้ง
- การวินิจฉัย DNA;
- ร่วมโปรแกรม;
- การทดสอบเหงื่อ.
ผู้ปกครองควรรู้: โรคที่ได้รับการวินิจฉัยก่อนหน้านี้ยิ่งมีโอกาสที่จะป้องกันการพัฒนาของโรคได้มากขึ้นการเปลี่ยนแปลงในสมอง.
คำแนะนำสำหรับการรักษา

จนถึงตอนนี้ในรัสเซียการรักษาด้วยอาหารยังคงเป็นเพียงผลกระทบของการรักษา. เภสัชวิทยาพยายามที่จะสร้างยาที่จะอนุญาตโดยไม่มีอาหารเพื่อควบคุมปริมาณของฟีนิลลาลันเนนในเลือด. ความสำเร็จในการทำงานของพวกเขาคือ แต่ยาเสพติดในการขายจะไม่ปรากฏเร็วกว่าหกปี.
นักวิทยาศาสตร์ยังคงมองหาวิธีการใหม่ในการจัดการพยาธิสภาพ.
- มีความหวังสูงสำหรับการรักษาเกษตรที่มีแนวโน้มเพราะยีนที่เปลี่ยนหายไปสามารถกำจัดปัญหาได้.
- ความพยายามที่จะใช้ Phenylalaninaliasis (เอนไซม์พืช) เพื่อให้มีส่วนร่วมในการแยกฟีนิลแมนน์ในร่างกายมนุษย์.
- ลองเข้าสู่เซลล์ของยีนของตับ Phenylalanninehydroxylase.
ในประเทศของเราจนถึงตอนนี้เด็กป่วยจะ จำกัด อาหารตั้งแต่แรกเกิดก่อนที่จะเกิดขึ้นของวัยแรกรุ่น (อายุ 16-18 ปี). สำหรับการพัฒนาและการเติบโตของผู้ป่วยดังกล่าวแพทย์หลายคนได้รับการสังเกตโดยเฉพาะกุมารแพทย์และนักประสาทวิทยา. ปรับจำนวนโปรตีนที่มาพร้อมกับอาหารอย่างต่อเนื่องเพื่อให้เข้ากับการโหลดและอายุ.
นอกจากนี้ยังมีรูปแบบของ FCU ซึ่งสามารถรับการรักษาด้วย tetrahydrobiodin. มันเป็นส่วนหนึ่งของเอนไซม์ที่เฉพาะเจาะจงที่ขาดอยู่ในร่างกายในโรคนี้.
เกี่ยวกับโภชนาการ Phenylketonuria

เพื่อปกป้องสมองและเซลล์ประสาทของเด็กจากอิทธิพลที่เป็นอันตรายของอนุพันธ์ของฟีนิลลานินินควรถูกลบออกจากอาหารของโปรตีนจากสัตว์. หากสิ่งนี้ทำในสัปดาห์แรกของชีวิตทารกแรกเกิดสมองจะยังคงอยู่ในความซื่อสัตย์ แต่ถ้าเวลาที่หายไปก็จะเป็นไปไม่ได้ที่จะกำจัดการเปลี่ยนแปลงที่เกิดขึ้นอย่างสมบูรณ์มันจะเป็นไปได้ที่จะระงับการเสื่อมสภาพต่อไป.
ผู้อ่านเว็บไซต์ของเราอาจมีคำถามที่สมเหตุสมผล: และวิธีที่ทารกจะเติบโตโดยไม่มีกรดอะมิโนที่มีอยู่ในโปรตีน? และกรดเหล่านี้จะมาที่ทารกในผลิตภัณฑ์ทางการแพทย์พิเศษ. ส่วนใหญ่มักจะผสมแห้ง. พวกเขาออกให้ผู้ปกครองฟรีในศูนย์พันธุศาสตร์.
หน้าอกได้รับมิกซ์ดังกล่าวที่ทำความสะอาดของแลคโตส.
องค์ประกอบของผลิตภัณฑ์โปรตีนประกอบด้วย:
- แยกโปรตีนนมแล้ว;
- กรดอะมิโนในรูปแบบฟรี (Taurine, Tyrosine, Tryptophan, Histidine, Cystine).
ตัวอย่างเช่นอาจเป็นส่วนผสม «apolylak», «ฟีนิลฟอง 1», «lofenalak», เช่นเดียวกับ «aponti», «Minafen», «ซิมารัน», «p-am» (และตัวเลข), «isifene» (นี่พร้อมใช้งานผลิตภัณฑ์).
ส่วนผสมของผงดังกล่าวควรได้รับการต้มด้วยน้ำต้มหรือผู้หญิงที่เขียนขึ้นก่อนหน้านี้.
มันได้รับอนุญาตให้ใส่น้ำนมแม่ให้กับทารก แต่แม่ของเขาจะต้องปฏิบัติตามอาหารพิเศษ.
เมื่อเด็กมาถึงเด็กก่อนวัยเรียนและวัยเรียนเขาไม่สามารถให้ผลิตภัณฑ์โปรตีนธรรมดา. ผลไม้, ผัก, น้ำมันพืช, ผลิตภัณฑ์แป้ง, วงกลมไม่มี.
เมื่อคุณทำเมนูสำหรับวันนี้คุณใช้ตารางเฉพาะที่แสดงมาตรฐาน Phenylalanine สำหรับอายุที่แน่นอน.
เด็ก ๆ ให้วิตามินและแร่ธาตุที่จำเป็นต้องมี Pyridoxine, Thiamine, Riboflavin, กรดโฟลิก, แมกนีเซียม, แคลเซียม, เหล็ก.
มีตารางพิเศษที่ผลิตภัณฑ์ทั้งหมดแบ่งออกเป็นสามกลุ่ม:
- รายการสีเขียว - พวกเขาได้รับอนุญาตให้กินโดยไม่มีข้อ จำกัด
- รายการสีส้ม - สามารถใช้งานได้ แต่ในปริมาณน้อย
- Red List - ผลิตภัณฑ์เหล่านี้ได้รับการยกเว้นอย่างสมบูรณ์จากอาหาร (ไข่, เนื้อ, ปลา, คอทเทจชีส, ถั่ว).
มีกลุ่มผลิตภัณฑ์อีกสองกลุ่ม:
- น้ำซุปข้นผลไม้สำเร็จรูป.
- ผลิตภัณฑ์ที่ถูกบล็อกต่ำเทียม - พาสต้าขนมปังคุกกี้ออกแบบมาสำหรับพลังงานอาหาร.
เด็กมักจะทำการวิเคราะห์เลือดของกระเพาะอาหารว่างเปล่า (ในตอนเช้า) เพื่อกำหนดจำนวนฟีนิลแมนน์ในนั้น:
- อายุสูงสุดสามเดือน - การควบคุมรายสัปดาห์
- จาก 3 ถึง 12 เดือน - รายเดือน;
- จากหนึ่งปีถึงสามปี - พอทุกสองเดือน
- จากนั้นก็อนุญาตให้บริจาคเลือดหนึ่งครั้งในสามเดือน.
ควรจำไว้ว่าเป็นไปไม่ได้ที่จะดื่มเครื่องดื่มอัดลมโดยเฉพาะกับ Phenylalanine (แอสปาร์แตม). ให้แน่ใจว่าได้อ่านองค์ประกอบของยาเสพติดที่ด้านหน้าของพวกเขาบางครั้งในพวกเขายังมีสารให้ความหวาน.
บางครั้งยาที่ปรับปรุงการไหลเวียนของสมองเช่น Nootropyl, Phenotropyl, Cerebrolysin, encephol. ในการปรับปรุงจุลภาค Pentoxifillain (Trental) จะถูกเขียนออกมา.
เด็กแนะนำให้นวด, แบบฝึกหัดยิมนาสติก, การรักษาพลศึกษา.
โรคทางพันธุกรรมนี้พัฒนาอย่างรวดเร็วสามารถนำไปสู่การเปลี่ยนแปลงที่ไม่สามารถย้อนกลับได้อันเป็นผลมาซึ่งเด็กสามารถกลายเป็นคนพิการได้เพิ่มไขมันลง. แต่ถ้าการวินิจฉัยเกิดขึ้นในสัปดาห์แรกของชีวิตและมีการปฏิบัติตามอาหารที่พัฒนาขึ้นเป็นพิเศษโดยไม่มีฟีนิลลานินินการพัฒนาของโรคจะหยุด - เด็กจะเติบโตมีสุขภาพดีจะได้รับการศึกษาอาชีพ. คนดังกล่าวมีครอบครัวของตัวเองพวกเขาสามารถเกิดเด็กปกติได้.